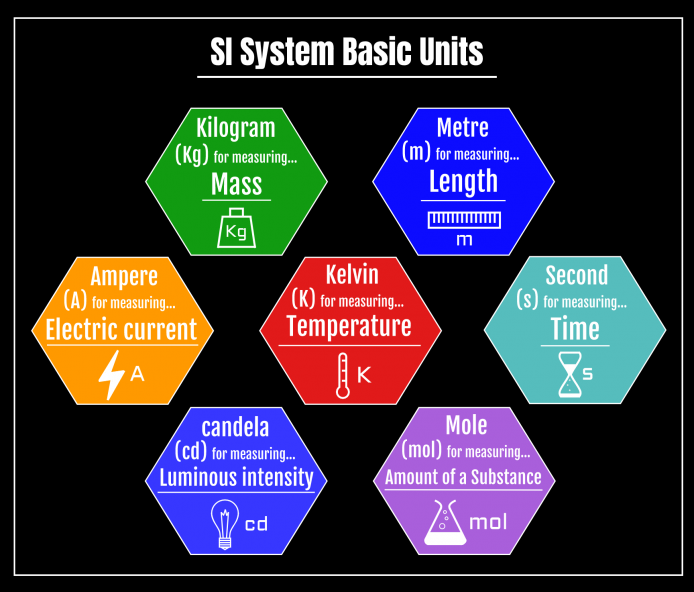
- Kilogram (kg), for mass
- Metre (m), for length
- Ampere (A), for electric current
- Kelvin (K), for temperature
- Second (s), for time
- Candela (cd), for luminous intensity
- Mole (mol), for amount of a substance
Another common unit to measure molecular mass is the Dalton (Da), which is defined as the mass of one hydrogen atom. This is commonly used in biology to describe the masses of amino acids and proteins. For example a protein with a molecular weight of 36,000 g mol-1, has a mass of 36,000 daltons, or 36 kDa.We also have other units that are defined in terms of these basic units, such as:
- Metres per second (m/s, ms-1) for speed
- Metres per second per second (m/s2, ms-2) for acceleration
- Pascals (Pa) for pressure, defined as the force of 1 N acting on an area of 1 m2
- Joules (J) for energy, defined as the work done by 1 N acting over a distance of 1 m.
We can often work out what the units for a physical quantity are by looking at the at the equation to calculate them. For example, speed is calculated as:\[\text{speed} = \frac{\text{distance}}{\text{time}} \sim \frac{\text{metres}}{\text{seconds}}\]So the units for speed are metres divided by seconds, or m/s. We could also write it as metres multiplied by seconds-1, or ms-1. We pronounce m/s, as metres per second. In general, whenever you see a "/" in a unit, you can replace it with "per".Similarly, we can calculate acceleration:\[\text{acceleration} = \frac{\text{speed}}{\text{time}} = \frac{\frac{\text{distance}}{\text{time}}}{\text{time}} = \frac{\text{distance}}{\text{time}^2} \sim \frac{\text{metres}}{\text{seconds}^2}\]Hence, acceleration is measured in metres divided by seconds2, or m/s2. Again, we could write it as metres multiplied by seconds-2, or ms-2. Or we could just say metres per second2, or metres per second per second.We often want to measure quantities that would give really big or really small numbers if we used the standard units for that quantity. Therefore it's convenient to have a system of prefixes that defines units of different orders of magnitude of the same unit, such as millimetres, centimetres, metres, kilometres etc.
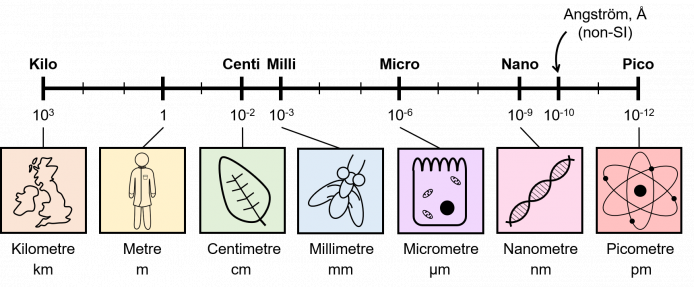
| Prefix | Multiplied by | Symbol |
|---|---|---|
| exa | \(10^{18}\) | E |
| peta | \(10^{15}\) | P |
| tera | \(10^{12}\) | T |
| giga | \(10^{9}\) | G |
| mega | \(10^6\) | M |
| kilo | \(10^3\) | k |
| hecto | \(10^2\) | h |
| deca | \(10\) | dk |
| deci | \(10^{-1}\) | d |
| centi | \(10^{-2}\) | c |
| milli | \(10^{-3}\) | m |
| micro | \(10^{-6}\) | \(\mu\) |
| nano | \(10^{-9}\) | n |
| pico | \(10^{-12}\) | p |
| femto | \(10^{-15}\) | f |
| atto | \(10^{-18}\) | a |
The table above shows the standard SI prefixes, along with the factor that you have to multiply the standard units by, and their symbols. For example, there are \(10^9\) metres in a gigametre, and a gigametre is denoted by Gm.To move up the table, you normally divide by \(10^3(=1000)\), and to move down the table you normally multiply by \(10^3(=1000)\). For example, to convert millimetres to metres, you divide by \(1000\), and to convert metres back to millimetres, you multiply by \(1000\). Note: This rule doesn't work to convert between hecto, deca, deci, or centi.
]]>- Mass (M)
- Length (L)
- Time (T)
- Electrical current (l)
- Temperature (\(\theta\))
Dimensions are not the same as units, for example we can measure speed in metres per second, kilometres per hour, or miles per hour; but it is always a length divided by a time regardless of the units, so the dimensions of speed are L/T.Similarly, we can measure area as square metres, or square kilometres, or square miles, but it is always a length squared, so the dimensions of area are L2.We can use this to work out the units of a constant or variable in an equation, by substituting the dimensions of the variables into the equation. This is based on the principle of consistency of dimensions, if two expressions are equal, their dimensions must be the same.Notation: To denote the dimensions of a physical quantity \(X\), we use square brackets, \([X]\).
]]>We know that force is mass times acceleration, \(F = ma\), so we can write the dimensions of force as \(\color{blue}{[F] = [m][a] = M L T^{-2}}\) since acceleration is a length divided by a time squared. We also have the dimensions of speed as a length over a time, \(\color{red}{[v] = L/T = LT^{-1}}\).
So now we can put this into our original equation:\[\color{blue}{MLT^{-2}} =[k](\color{red}{LT^{-1}})^2 = [k]L^2T^{-2}\]Now we rearrange to find \([k]\).\[\begin{align} [k] &= \frac{MLT^{-2}}{L^2T^{-2}} \\ &= \frac{ML}{L^2}\\ &=\frac{M}L \end{align}\]So we now that \(k\) is a mass divided by a length, or we can measure it in terms of kg/m.
]]>It's helpful to write down all the dimensions we know:
- Force is \(\color{blue}{MLT^{-2}}\).
- Radius is a length, \(\color{red}L\).
- Length is a length, \(\color{green}L\).
- Speed is a length divided by a time, \(\color{purple}{LT^{-1}}\).
- Distance is a length, \(\color{orange}L\).
We can now substitute all of these into the equation, noting that we can ignore the dimensions of \(-2\pi\) since that's just a number that has no dimensions.\[\begin{align} \color{blue}{MLT^{-2}} &= \color{red}L \color{green}L \frac{\color{purple}{LT^{-1}}}{\color{orange}L}[\eta] \\ &= L^2T^{-1}[\eta] \\ MT^{-2} &= LT^{-1}[\eta] \\ L[\eta] &= MT^{-1} \\ [\eta] &= ML^{-1}T^{-1} \end{align}\]So we have that the dimensions of viscosity are a mass divided by a length and a time, so we can measure it in kgm-1s-1.
]]>Suppose that \(v\) is proportional to some power of \(l\), that \[v \propto l^a\]Use dimensional arguments to find \(a\).
Since \(v\) depends on \(l\) and \(g\), we can write\[v = Cl^a g^b\]for some dimensionless constant \(C\). Now taking dimensions gives us\[LT^{-1} = L^a(LT^{-2})^b = L^{a + b}T^{-2b}\]since \(g\) is an acceleration due to gravity. Matching the dimensions yields\[1 = a+ b \\ -1 = -2b\]which can be solved to give\[b = \frac12 \\ a = \frac12\](So this gives \(v \propto \sqrt{gl}\).)
]]>Taking dimensions of the equation gives us\[T = M^a(ML^{-3})^b(L^2T^{-1})^c = M^{a+b}L^{-3b+2c}T^{-c}\]which yields the equations\[1 = -c \\ 0 = a + b \\ 0 = -3b+2c\]Solving these simultaneously gives us\[a = \frac23 \\ b =-\frac23 \\ c = -1\]So then\[t = Cm^{\frac23}\rho^{-\frac23}\kappa^{-1} \propto m^{\frac23}\rho^{-\frac23}\kappa^{-1}\]
]]>- \[\sqrt{\frac{3RT}{M}}\]
- \[\sqrt{\frac{M}{3RT}}\]
Based on the observation that the terminal velocity \(v\) is proportional to the powder density, express \(v\) in terms of \(\rho\), \(g\), \(d\), and \(\eta\) using dimensional analysis.
]]>So now we have\[LT^{-1}= M^{1+z}L^{x+y+z-3}T^{-2x-z}\]
Matching the LHS and RHS dimensions yields\[z=-1 \\ x+ y - z - 3 = 1 \\ -2x+1 = -1\]Solving the simultaneous equations yields\[x=1 \\y=2\\z=-1\]
Thus we obtain \[v = \Gamma\rho g d^2\eta^{-1}\]
Experiments show that \(R\) is proportional to the length of the blood vessel \(L\) (m). Use dimensional analysis to show that \(R\) is expressed in terms of \(L\), the viscosity of blood (Pa s), and the radius of the blood vessel \(r\) (m) in the following form:
\[R = \Gamma \frac{L\eta}{r^4}\]
where \(\Gamma\) is a dimensionless constant.
]]>\[R = \Gamma L \eta^xr^y\]
where \(\Gamma\) is a unit-less constant. The dimensions are
Left hand side: \[[\text{Pa}] L^{-3}T= ML^{-1}T^{-2}L^{-3}T = ML^{-4}T^{-1}\]
Right hand side: \[L([\text{Pa}]T)^xL^y = L(ML^{-1}T^{-1})^xL^y = L^{1-x+y}M^xT^{-x}\]Consistency of units requires
\[x=1 \\1-x+y=-4\]
Solving the simultaneous equations yields
\[x=1\\y=-4\]
Thus we obtain
\[R=\Gamma L\eta r^{-4} = \Gamma\frac{L\eta}{r^4}\] ]]>
- \[\sqrt{\frac{3RT}{M}}\]
- \[\sqrt{\frac{M}{3RT}}\]
Charged molecules (each with charge \(q\) [C=coulomb]) travel through the electric field \(E\) (N C-1). Each molecules experience frictional drag \(f\) (N s m-1).
Calculate the speed \(v\) of the molecule.
(a) Express the flight time \(t\) (s) in terms of mass of the ion \(m\) (kg), charge of the ion \(q\) (C = coulomb), the electrostatic potential \(\varepsilon\) (J C-1), and the length of flight \(L\).
(b) Express the speed of the ion \(v\) in terms of the mass \(m\), charge \(q\) and the electrostatic potential \(\varepsilon\).
(c) Does your answer make sense?
]]>
.png')
.png')
.png')
.png') ]]>
]]>.png') ]]>
]]>![Scatter plot of fractional saturation vs [L]](FileLocation + 'media/image(93).png') ]]>
]]>.png') ]]>
]]>.png') ]]>
]]>.png') ]]>
]]>![Scatter plot of fractional saturation vs [L]](FileLocation + 'media/image(94).png') ]]>
]]>.png') ]]>
]]>




.png')
.png')
.png')
.png')
.png')
.png')
.png')
.png')
.png')
.png')
.png')







.png')
.png')
.png')



.png')
.png')


.png')

.png')


.png')

